Basecalling
Instructors
- Nick Gleadall (ng384@medschl.cam.ac.uk)
- Alba Sanchis Juan (as2635@medschl.cam.ac.uk)
1) Aims
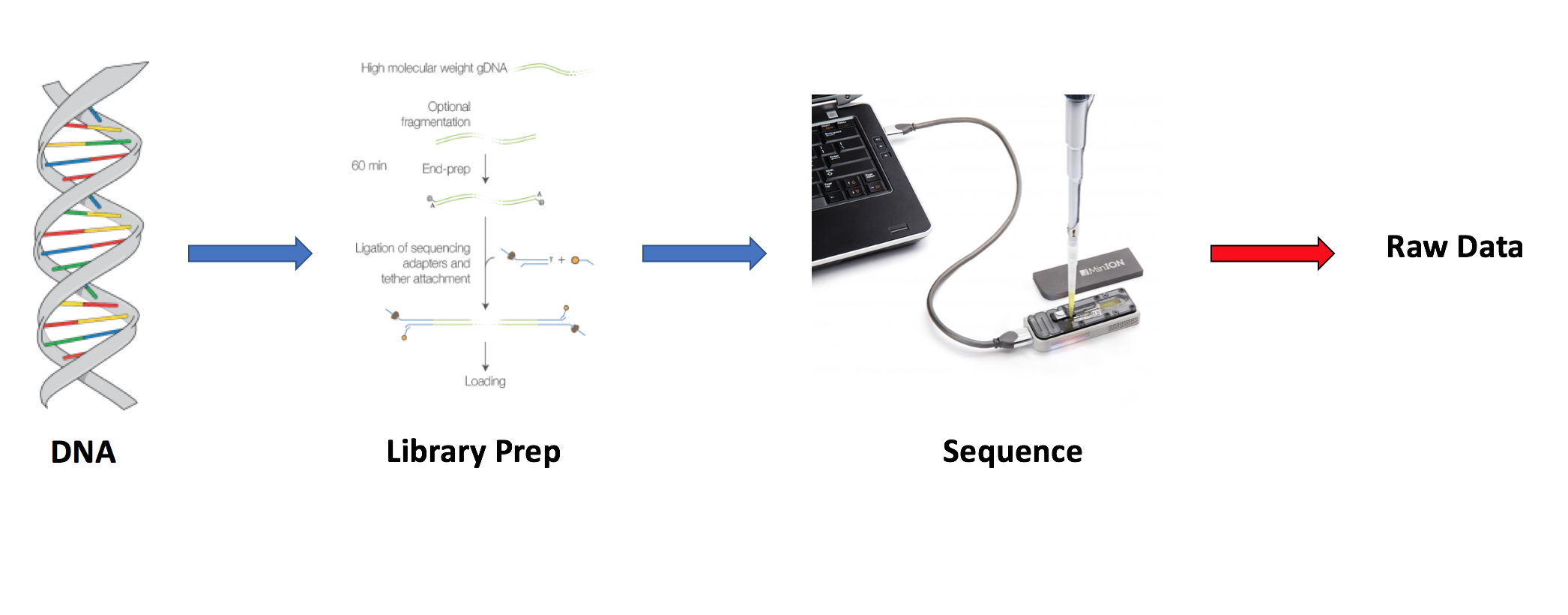
Overall: How to take Oxford Nanopore (ON) data from raw signal to called variants
In this part of the session we will learn about Data formats , Basecalling and Basic Quality Control(QC)
2) Starting point
3) Raw reads
All nanopore data is written to a specific run directory. Change to the example directory below…
cd /mnt/albasj/data/nanopore/
… and list it to see what is there
ls
Lets take a look in the reads/ directory. This is where the sequencer writes raw data.
ls reads/
You should see two directories: (0/ and 1/)
ON machines write reads in batches (here batch size is 4000) This is to keep the number of files in each directory resonable for the computers filesystem
ls reads/0/
4) FAST5 files
Nanopore writes read data to a file format they call FAST5
1 read = 1 .fast5 file
FAST5 files are infact HDF5 files. These are compressed binary files which store data in a structured way, allowing fast random access to subsets of the data.
This is where electronic signal data from the sequencer is storred
Lets look at the structure of a FAST5 file using h5ls
h5ls reads/0/GXB01206_20180518_FAH88225_GA50000_sequencing_run_CD3_92236_read_9998_ch_295_strand.fast5
This shows the top level data keys.
We can view the subkeys by recursivley listing the file
h5ls -r reads/0/GXB01206_20180518_FAH88225_GA50000_sequencing_run_CD3_92236_read_9998_ch_295_strand.fast5
We can also dump and view the entire contents of a FAST5
h5dump reads/0/GXB01206_20180518_FAH88225_GA50000_sequencing_run_CD3_92236_read_9998_ch_295_strand.fast5 | less
(Hint 1: use the mouse wheel to scroll up and down the file) (Hint 2: press q to exit less, a text reading program)
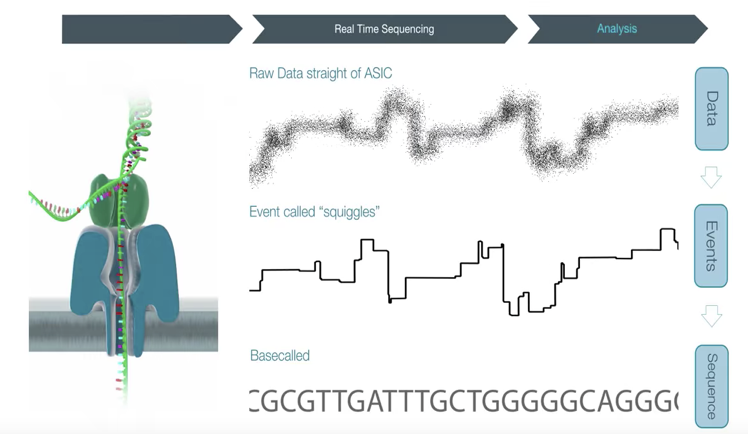
5) Basecalling
This is the process of translating raw electrical signal data from an ON sequencer to DNA sequence. Basecalling is a critical step in the analysis workflow as poor basecalling makes poor sequence data.
Many basecallers exist - but for now we will be using Albacore v2.3.3 developed by ON
For basecalling it is important to know which Flow Cell and Library Prep Kit was used. To see all the combinations which Albacore can handle try:
read_fast5_basecaller.py -l
So lets basecall
username=_yourusername_
read_fast5_basecaller.py --flowcell FLO-MIN106 \
--kit SQK-PCS108 \
--input reads/ \
--recursive \
--worker_threads 4 \
--save_path /mnt/albasj/analysis/${username}/basecalled_reads/
(Interesting fact: You have just started up a Machine Learning script. Albacore, alongside almost all current nanopore basecallers have a neural network at their core)
5.i) A comment about basecallers
As previously mentioned many basecallers are available.
The main performance marker of a basecaller that we care about is the overall Assembly Identity (how much a final alignment matches the reference)
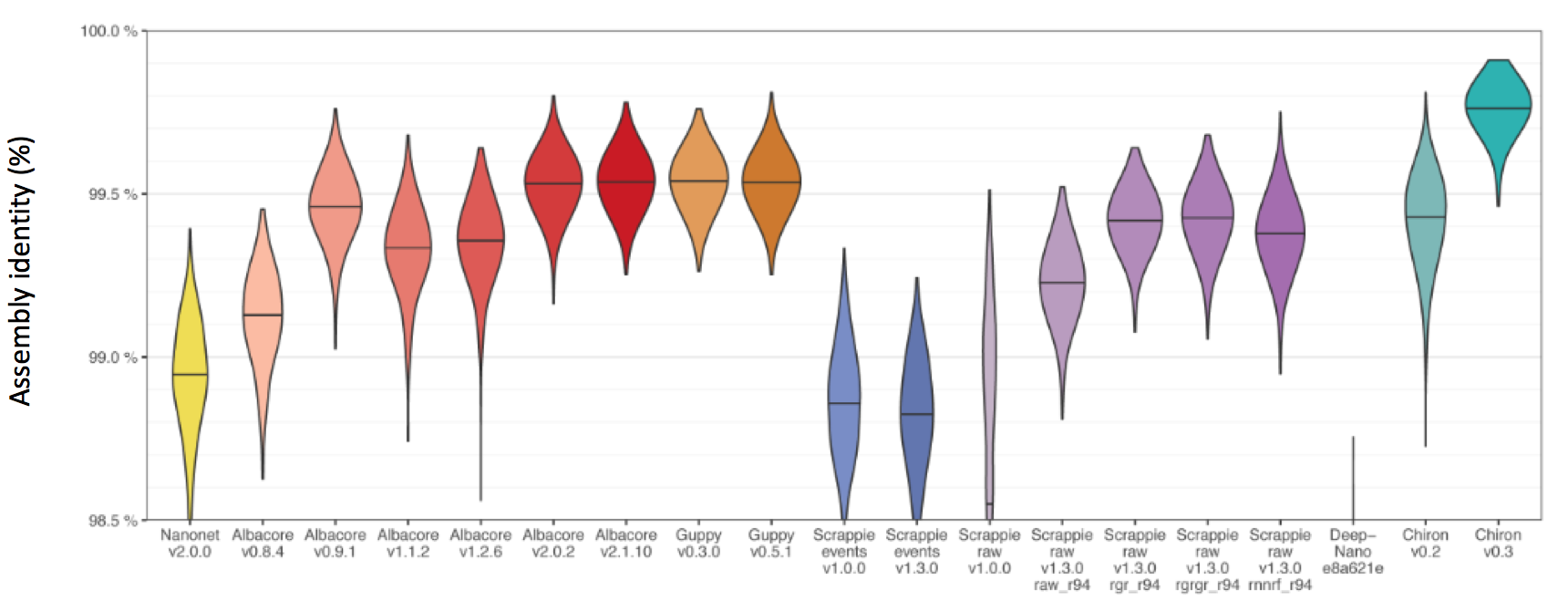
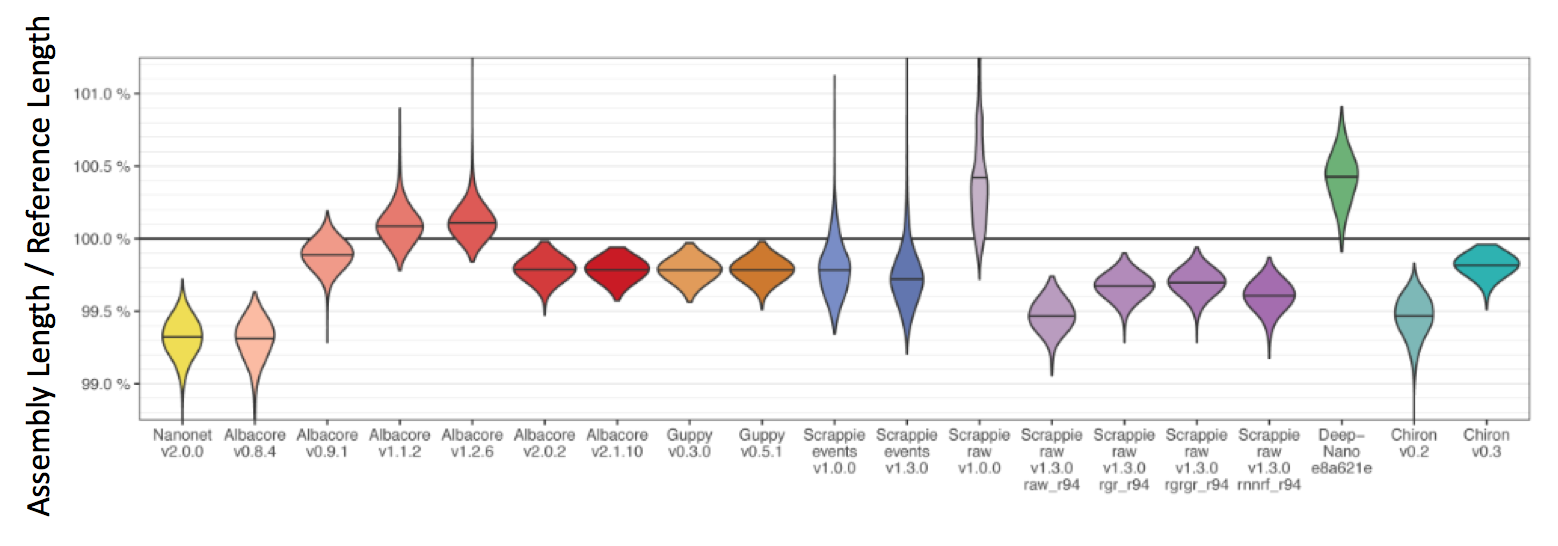
We can also take a look at the assembly length bias which tells us if a given basecaller is prone to reference insertions or deletions
(Further reading: A great comparison of basecallers exists here: basecaller_comparison
6) Recommended workflow
There are two different workflows we recommend based on which sequencer you have available.

7) Basecalling results
Lets take a look inside the new basecalled_reads/ directory we just created
cd /mnt/albasj/analysis/${username}
ls -1 basecalled_reads/
Important directory: workspace/
ls basecalled_reads/workspace/
This contains two sub-directories pass/ and fail/ which contain the basecalled read data in fastq format (ready for use with common aligners like BW).
pass/ obviously contains all of the reads which can be used in further downstream analysis and fail/ contains reads which are un-useable
ls basecalled_reads/workspace/pass/
To see the basic format of a .fastq file run the command below. fastq’s will be covered further in tomorrows session
less basecalled_reads/workspace/pass/fastq_runid_eeb92128ecdaf98ba8cd29e26976e99b3843f88e_0.fastq
(Hint: press q to exit less, a text reading program)
Important file: sequencing_summary.txt
less basecalled_reads/sequencing_summary.txt
(Hint: press q to exit less, a text reading program)
This file can be used to plot basic run statistics as part of QC
8) Basic run QC
It is much easier to view this data in R.
Open R:
R
and execute the following commands:
library(ggplot2)
summary_data <- read.table( "basecalled_reads/sequencing_summary.txt", sep="\t", header=TRUE)
ggplot(data=summary_data, aes(sequence_length_template)) +
geom_histogram()
You can also represent the length template by qscore template:
ggplot(data=summary_data, aes(x=mean_qscore_template, y=sequence_length_template, color=passes_filtering)) +
geom_point()
Hint: to quit R, type: quit()
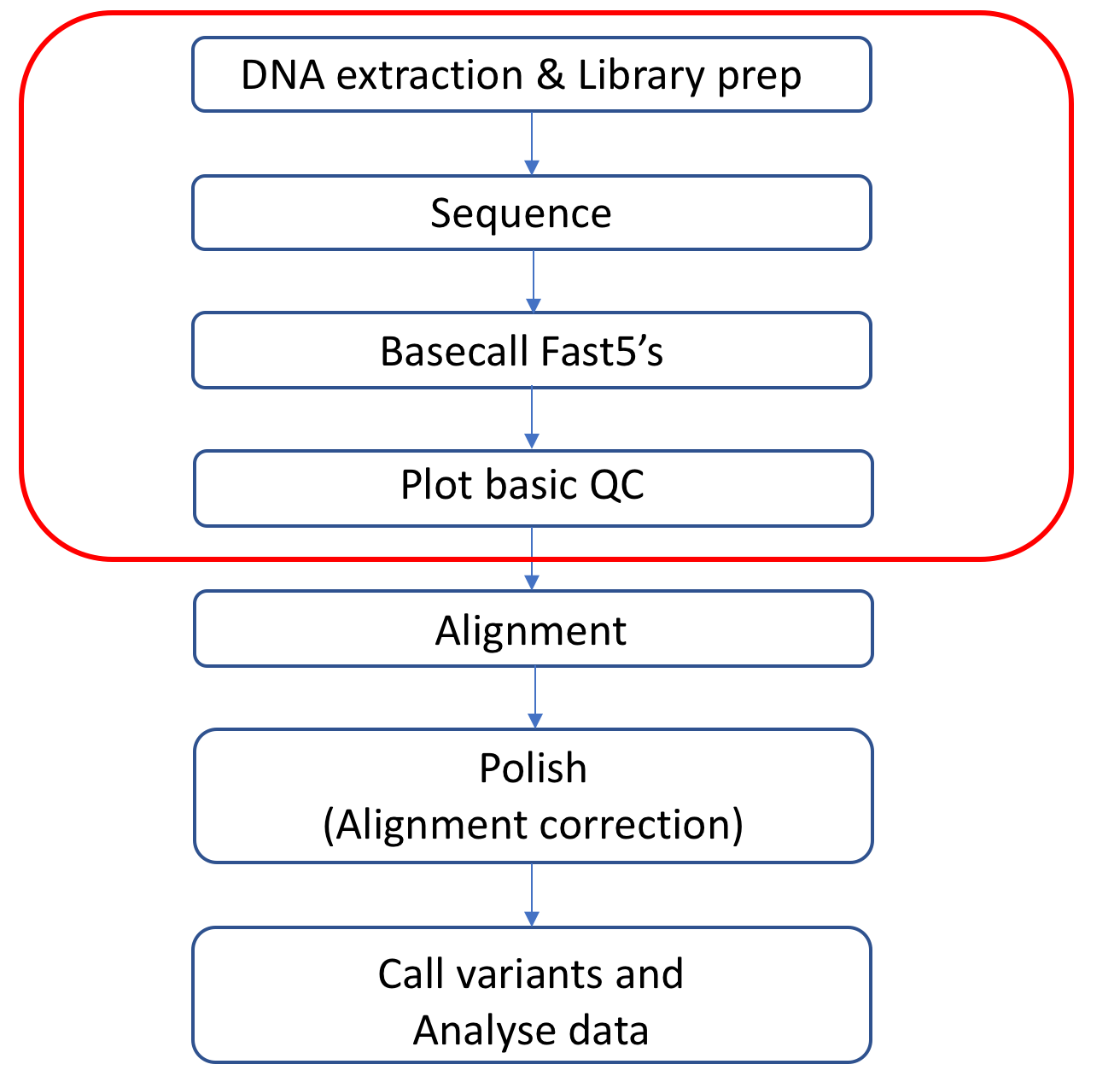
Final Comment
This was a VERY basic overview of nanopore data analysis. Below is a diagram showing the parts of an overall workflow this tutorial has covered.